Tutorial 4: running convergence tests
While koopmans is a package primarily oriented towards performing Koopmans functional calculations, it does have a couple of other useful functionalities. Among these functionalities is the ability to perform arbitrary covnergence tests.
A simple convergence test
In this tutorial, we will make use of its convergence task to determine how large a cell size and energy cutoff is required to converge the PBE energy of the highest occupied molecular orbital (HOMO) of a water molecule. This workflow was chosen for its simplicity; it is possible to run convergence tests on any workflow implemented in the koopmans package.
The input file
In order to run this calculation, in the workflow block we need to set the converge parameter to true:
{
"workflow": {
"functional": "dft",
"task": "singlepoint",
"converge": true,
"fix_spin_contamination": false,
"pseudo_library": "SG15/1.0/PBE/SR"
},
"engine": {
and then provide the convergence information in the convergence block:
"from_scratch": true
},
"convergence": {
"observable": "homo energy",
"threshold": "0.01 eV",
"variables": [
"ecutwfc",
"celldm1"
These settings state that we are going to converge the HOMO energy to within 0.01 eV, with respect to both the energy cutoff ecutwfc and the cell size. The full input file can be found here.
The output file
When you run the calculation, you should see something like this after the header:
Singlepoint ecutwfc 20_0 celldm1 11_3
DFTCP
✅
01-dftcompleted
Singlepoint ecutwfc 20_0 celldm1 12_3
DFTCP
✅
01-dftcompleted
Singlepoint ecutwfc 20_0 celldm1 13_3
DFTCP
✅
01-dftcompleted
Singlepoint ecutwfc 30_0 celldm1 11_3
DFTCP
✅
01-dftcompleted
Singlepoint ecutwfc 30_0 celldm1 12_3
DFTCP
✅
01-dftcompleted
Singlepoint ecutwfc 30_0 celldm1 13_3
DFTCP
✅
01-dftcompleted
Here, the code is attempting to use progressively larger energy cutoffs and cell sizes. It will ultimately arrive at a converged solution, with a ecutwfc of 50.0 Ha and a cell slightly larger than that provided in the .json input file.
Plotting
The individual Quantum ESPRESSO calculations reside in nested subdirectories. If you plot the HOMO energies from each of these, you should get something like this:
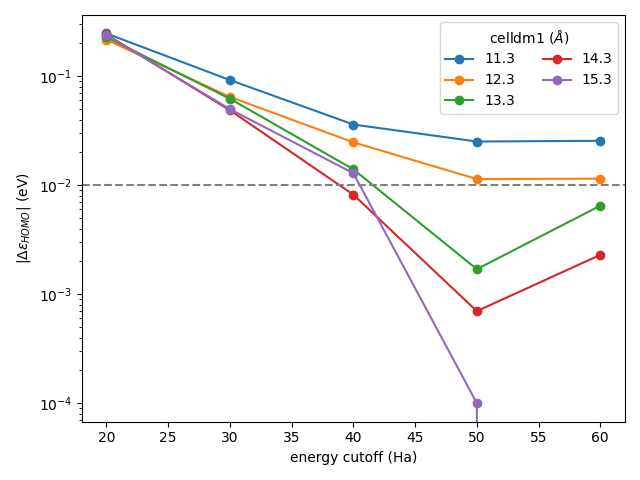
Plot of the HOMO energy of water with respect to the energy cutoff and cell size (generated using this script)
We can see that indeed the calculation with ecutwfc = 50.0 and celldm(1) = 13.3 is the one where the energy of the HOMO goes within (and stays within) 0.01 eV of the most accurate calculation.
A custom convergence test
In the previous example, we performed a convergence test with respect to ecutwfc and celldm1. A full list of supported convergence variables can be found here. You will see that only a couple of variables are implemented by default. But don’t let that limit you! It is possible to perform a convergence test on arbitrary keywords using factories.
First, try taking the input file from the first part of the tutorial, and switch the pseudopotential library to pseudo_dojo_standard. What do you notice?
Hopefully, the first thing you will see is that there are now some warnings about the small box parameters nrb like this,
🚨 Small box parameters `nrb` not provided in input: these will be automatically set to safe default values. These
values can probably be decreased, but this would require convergence tests. Estimated real mesh dimension `(nr1,
nr2, nr3) = 36 36 45`. Small box mesh dimension `(nr1b, nr2b, nr3b) = 20 18 20`.
These parameters are associated with the way Quantum ESPRESSO handles non-local core corrections in pseudopotentials, and corrections are present in the new set of pseudopotentials but absent in the SG15 pseudopotentials.
So, let’s perform a convergence test! The nrb parameters are not directly implemented as a convergence variable in koopmans, but we can use a factory to perform a convergence test on them, by making use of the ConvergenceVariable and ConvergenceWorkflowfactory classes.
"""A simple script that converges the HOMO energy of a water molecule with respect to nr1b, nr2b, and nr3b."""
from ase_koopmans.build import molecule
from koopmans import engines, workflows
# Initialize the engine to run the workflow
engine = engines.LocalhostEngine()
# Use ASE to construct a water molecule
atoms = molecule('H2O', vacuum=5.0)
# Create a subworkflow which calculates (among other things) the PBE HOMO energy of water
subworkflow = workflows.DFTCPWorkflow(atoms=atoms, ecutwfc=30.0, base_functional='pbe',
pseudo_library='PseudoDojo/0.4/PBE/SR/standard/upf',
calculator_parameters={'kcp': {'nr1b': 6, 'nr2b': 6, 'nr3b': 6}},
engine=engine)
# koopmans doesn't implement convergence with respect to nrb, so we need to define a custom
# ConvergenceVariable. To do so, we must first...
# ... define a function that extracts nr1-3b from a workflow
def get_nrb(workflow):
"""Get the values for nr1-3b from a workflow."""
return [workflows.get_calculator_parameter(workflow, f'nr{i}b') for i in [1, 2, 3]]
# ... define a function that sets nr1-3b
def set_nrb(workflow, value):
"""Set nr1-3b equal to `value` for a workflow."""
workflows.set_calculator_parameter(workflow, 'nr1b', value[0])
workflows.set_calculator_parameter(workflow, 'nr2b', value[1])
workflows.set_calculator_parameter(workflow, 'nr3b', value[2])
nrb_variable = workflows.ConvergenceVariable(name='nrb',
increment=[2, 2, 2],
get_value=get_nrb,
set_value=set_nrb)
# Create the convergence workflow using the convergence factory
workflow = workflows.ConvergenceWorkflowFactory(subworkflow,
observable='total energy',
threshold=1e-3,
variables=[nrb_variable])
# Run the workflow
workflow.run()
Running this script will perform a convergence test with respect to nrb 1-3.
Warning
This tutorial performs convergence tests in a slightly incorrect way, To see this, add the keyword length = 10 to the ConvergenceVariable in the above script. How is the behavior different? Which behavior is correct? Why?